This page was considered obsolete and made private on March 2016, and is not accessible to the public anymore.
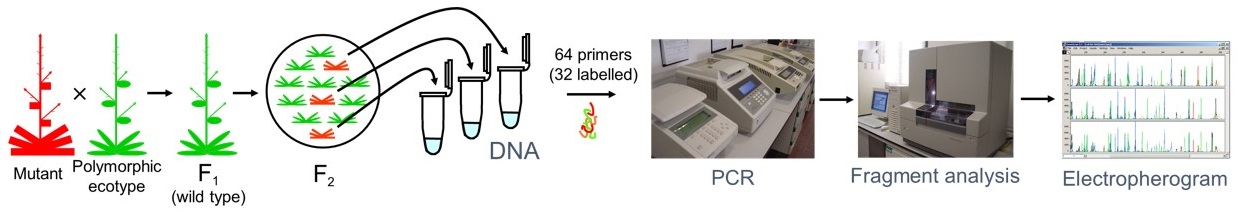
Map-based cloning is a classic approach for the molecular identification of a gene when a molecular tag is not available. With the completion of the genome sequence and the availability of enough molecular markers to cover the entire genome, map-based cloning emerged as a powerful approach to identifying genes altered by mutation in many experimental organisms, including plant species other than Arabidopsis. We developed a high-throughput mapping procedure based on simultaneous PCR coamplification, and fluorescent semiautomated detection and sizing of several microsatellite markers (Ponce et al., 1999). Once the F2 mapping population is ready, less than 1 week is required for low-resolution mapping and less than 1 month for delimiting a 100-200 kb candidate interval with our method (Ponce et al., 2006). The use of this approach allowed us to assign map positions to 98 of the genes identified by mutation in our laboratory, including 86 induced by EMS, nine taken from the AIS collection, one induced by fast neutrons, and one of spontaneous origin (Robles and Micol, 2001; Micol, 2009; Pérez-Pérez et al., 2009).
The map positions determined with our high-throughput method have led to the positional cloning of genes identified in our screens. This is the case for 59 of the 98 mutations we initially mapped. The products of these genes participate in various developmental processes, such as polar cell expansion, transduction of hormonal signals, gene regulation, plastid biogenesis, and chromatin remodelling, among others. The range of phenotypes and processes identified reveal the complexity of leaf ontogeny and will help explain the diversity of leaf morphology in nature.
Massively parallel sequencing technologies (Next Generation Sequencing; NGS) have enabled the rapid cloning of genes identified in forward genetic screens. We found that resequencing the genomes of fine mapped mutants is a feasible approach that leads to the identification of a moderate number of mutations within the candidate interval, including the one that causes the mutant phenotype (Mateo-Bonmatí et al., 2014; Candela et al., 2015).
If you are interested in our mapping and cloning service (Arabidopsis point mutations only), please email Prof. Ponce.